
First-principles calculations and experimental studies of XYZ2 thermoelectric compounds: detailed analysis of van der Waals interactions†

Abstract
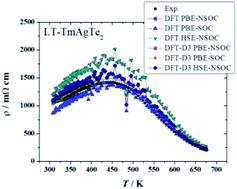
First-principles calculations can accelerate the search for novel high-performance thermoelectric materials. However, the prediction of the thermoelectric properties is strongly dependent on the approximations used for the calculations. Here, thermoelectric properties were calculated with different computational approximations (i.e., PBE-GGA, HSE06, spin–orbit coupling and DFT-D3) for three layered XYZ2 compounds (TmAgTe2, YAgTe2, and YCuTe2). In addition to the computations, the structural, electrical and thermal properties of these compounds were measured experimentally and compared to the computations. An enhanced prediction of the crystal structure and heat capacity was achieved with the inclusion of van der Waals interactions due to more accurate modeling of the interatomic forces. In particular, a large shift of the acoustic phonons and low-frequency optical phonons to lower frequencies was observed from the dispersion-optimized structure. From the phonon dispersion curves of these compounds, the ultralow thermal conductivity in the investigated XYZ2 compounds could be described by a recent developed minimum thermal conductivity model. For the prediction of the electrical conductivity, a temperature-dependent relaxation time was used, and it was limited by acoustic phonons. While HSE06 has only a small influence on the electrical properties due to a computed band gap energy of >0.25 eV, the inclusion of both van der Waals interactions and spin–orbit coupling leads to a more accurate band structure, resulting in better prediction of electrical properties. Furthermore, the experimental thermoelectric properties of YAgTe2, TmAg0.95Zn0.05Te2 and TmAg0.95Mg0.05Te2 were measured, showing an increase in zT of TmAg0.95Zn0.05Te2 by more than 35% (zT = 0.47 ± 0.12) compared to TmAgTe2.
 Please wait while we load your content...
Please wait while we load your content...

