
Tailoring chemical expansion by controlling charge localization: in situ X-ray diffraction and dilatometric study of (La,Sr)(Ga,Ni)O3−δ perovskite†
Abstract
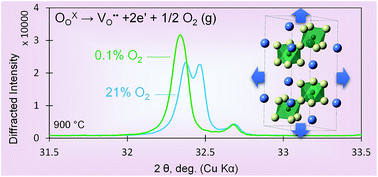
Large coupling of the oxygen content and lattice dilation, known as chemical expansion, deleteriously impacts the durability of non-stoichiometric active oxides in energy conversion and storage devices. In this work, the role of charge localization on multivalent cations during reduction was experimentally probed using La0.9Sr0.1Ga1−xNixO3−δ (LSGN; x = 0.05, 0.1, and 0.5) to test prior theoretical predictions of decreased chemical expansion upon decreasing charge localization. Increasing the Ni content in LSGN resulted in a decrease in the coefficient of chemical expansion, consistent with expected charge de-localization as LSGN transitions from polaron hopping to metallic conduction, as demonstrated in prior electrical studies. Dilatometry and thermogravimetry showed that samples with more Ni underwent larger changes in the oxygen content and consequently expanded more, though the normalized expansion for a given stoichiometry change (the coefficient of chemical expansion) was less. In situ X-ray diffraction revealed an increase of crystal symmetry with a decreasing oxygen partial pressure and/or increasing temperature, consistent with expected changes in Ni and anion radii and demonstrating the interplay between chemical expansion and the crystal structure. The smaller-than-expected magnitude of the change in the chemical expansion coefficient with charge delocalization, compared to prior theoretical predictions, suggests that other factors, such as structural changes and the nature of the multivalent cation, likely also contribute. Finally, the effective radius of an oxygen vacancy (smaller than an oxide ion) as well as the change in size of Ni with charge de-localization was estimated.
 Please wait while we load your content...
Please wait while we load your content...

