
Resolving alternative organic crystal structures using density functional theory and NMR chemical shifts†‡
 a
James D.
Farrell,
b
Jason C.
Cole,
c
Judith A. K.
Howard
d
and
Paul
Hodgkinson
*d
a
James D.
Farrell,
b
Jason C.
Cole,
c
Judith A. K.
Howard
d
and
Paul
Hodgkinson
*d
Abstract
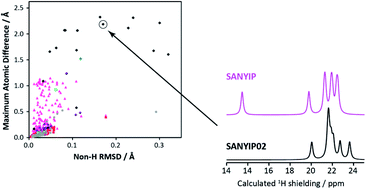
Alternative (‘repeat’) determinations of organic crystal structures deposited in the Cambridge Structural Database are analysed to characterise the nature and magnitude of the differences between structure solutions obtained by diffraction methods. Of the 3132 structure pairs considered, over 20% exhibited local structural differences exceeding 0.25 Å. In most cases (about 83%), structural optimisation using density functional theory (DFT) resolved the differences. Many of the cases where distinct and chemically significant structural differences remained after optimisation involved differently positioned hydroxyl groups, with obvious implications for the correct description of hydrogen bonding. 1H and 13C chemical shifts from solid-state NMR experiments are proposed as an independent methodology in cases where DFT optimisation fails to resolve discrepancies.
 Please wait while we load your content...
Please wait while we load your content...

