
Nontrigonal constraint enhances 1,2-addition reactivity of phosphazenes†

Abstract
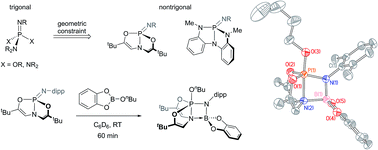
The syntheses and 1,2-addition reactivities of nontrigonal phosphazenes supported by trianionic tricoordinating chelates of the type L3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ndipp (3: L3 = N[CHC(tBu)O]23−; 4: L3 = N(o-NMeC6H4)23−; dipp = 2,6-diisopropylphenyl) are reported. These compounds are characterized by multinuclear NMR and single-crystal X-ray diffraction experiments. Distorted phosphazenes 3 and 4 are shown to add B–H, B–O, and Si–H bonds across the formal PN double bond, and their reactivities are contrasted with acyclic analogues. Derivatives of phosphazene 3 bearing sterically unencumbered N-substitutents readily dimerize to form the corresponding cyclodiphosphazanes; compounds with sterically demanding N-substituents are interconvertible between their monomeric and dimeric forms. The enhanced electrophilicity of the phosphorus center in nontrigonal phosphazenes 3 and 4 is rationalized by DFT calculations. Gas phase fluoride ion affinities are computed to be markedly higher for distorted phosphazenes, while proton affinities are largely unaffected by geometric distortion. These results are interpreted to suggest that distortion from pseudotetrahedral geometry results in stabilization of the P-based LUMO, while HOMO energies are essentially unchanged.
Ndipp (3: L3 = N[CHC(tBu)O]23−; 4: L3 = N(o-NMeC6H4)23−; dipp = 2,6-diisopropylphenyl) are reported. These compounds are characterized by multinuclear NMR and single-crystal X-ray diffraction experiments. Distorted phosphazenes 3 and 4 are shown to add B–H, B–O, and Si–H bonds across the formal PN double bond, and their reactivities are contrasted with acyclic analogues. Derivatives of phosphazene 3 bearing sterically unencumbered N-substitutents readily dimerize to form the corresponding cyclodiphosphazanes; compounds with sterically demanding N-substituents are interconvertible between their monomeric and dimeric forms. The enhanced electrophilicity of the phosphorus center in nontrigonal phosphazenes 3 and 4 is rationalized by DFT calculations. Gas phase fluoride ion affinities are computed to be markedly higher for distorted phosphazenes, while proton affinities are largely unaffected by geometric distortion. These results are interpreted to suggest that distortion from pseudotetrahedral geometry results in stabilization of the P-based LUMO, while HOMO energies are essentially unchanged.
 Please wait while we load your content...
Please wait while we load your content...

