
Quantum molecular modelling of ibuprofen bound to human serum albumin†
Abstract
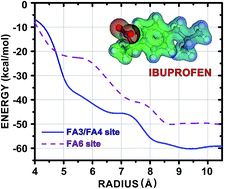
The binding of the nonsteroidal anti-inflammatory drug ibuprofen (IBU) to human serum albumin (HSA) is investigated using density functional theory (DFT) calculations within a fragmentation strategy. Crystallographic data for the IBU–HSA supramolecular complex shows that the ligand is confined to a large cavity at the subdomain IIIA and at the interface between the subdomains IIA and IIB, whose binding sites are FA3/FA4 and FA6, respectively. The interaction energy between the IBU molecule and each amino acid residue of these HSA binding pockets was calculated using the Molecular Fractionation with Conjugate Caps (MFCC) approach employing a dispersion corrected exchange–correlation functional. Our investigation shows that the total interaction energy of IBU bound to HSA at binding sites of the fatty acids FA3/FA4 (FA6) converges only for a pocket radius of at least 8.5 Å, mainly due to the action of residues Arg410, Lys414 and Ser489 (Lys351, Ser480 and Leu481) and residues in non-hydrophobic domains, namely Ile388, Phe395, Phe403, Leu407, Leu430, Val433, and Leu453 (Phe206, Ala210, Ala213, and Leu327), which is unusual. Our simulations are valuable for a better understanding of the binding mechanism of IBU to albumin and can lead to the rational design and the development of novel IBU-derived drugs with improved potency.
 Please wait while we load your content...
Please wait while we load your content...

