
Polyethenetetrathiolate or polytetrathiooxalate? Improved synthesis, a comparative analysis of a prominent thermoelectric polymer and implications to the charge transport mechanism†

Abstract
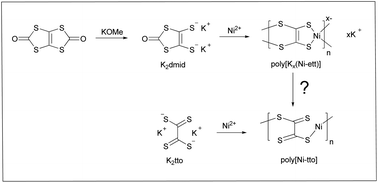
1,1,2,2-Ethenetetrathiolate (ett4−) coordination polymers, such as poly[Kx(Ni-ett)], have been known for decades for their excellent thermoelectric properties. However in reality, ett4− is neither a “true” comonomer which participates in the polymerization, nor represents a “true” repeat unit of the target polymer. Indeed, poly[K2(Ni-ett)], which is formally the product of Ni-induced polymerization of ett4−, has a poor conductivity and needs to be oxidized to show attractive thermoelectric characteristics. The polymerization and oxidation processes are poorly controllable which causes irreproducibility of the polymer properties. To improve the synthesis reproducibility, we studied polymerization of potassium tetrathiooxalate (K2tto), the convenient synthesis of which was developed in our recent work. Because K2tto is the “true monomer”, and not its precursor, a high quality product is reproducibly formed simply by mixing K2tto with NiCl2 at room temperature. The procedure does not require additional components (bases), or special conditions (prolonged heating), which are usually needed for the preparation of this polymer from the monomer precursor 1,3,4,6-tetrathiapentalene-2,5-dione (TPD). Furthermore, as tto2− is formally the product of two-electron oxidation of ett4−, the poorly controllable oxidation process is avoided and poly[Ni-tto] almost free from K is directly formed upon the complexation of Ni2+ and tto2−. Thus-obtained poly[Ni-tto] possesses conductivity in the range of 27–47 S cm−1 and a Seebeck coefficient in the range of −38 to −55 μV K−1, which are superior thermoelectric properties compared to poly[Kx(Ni-ett)] samples obtained by the previously reported methods. Redox and structural properties of poly[Ni-tto] were compared with those of poly[Kx(Ni-ett)] obtained by the reported methods. Furthermore, DFT calculations were performed to shed more light on generally promising properties of this class of materials. Particularly, possible packing models have been predicted for polymers, and the molecular dynamics simulations have been used to simulate the molecular arrangements under ambient conditions.
 Please wait while we load your content...
Please wait while we load your content...

