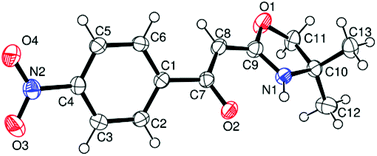
A synthetic, structural and theoretical investigation into the solid-state, solution and gas phase structure(s) of six 2-acylmethyl-4,4-dimethyl-2-oxazolines is reported. Four of these materials, viz. α-[(4,5-dihydro-4,4-dimethyl-2-oxazolyl)methylene]benzenemethanol (3a), α-[(4,5-dihydro-4,4-dimethyl-2-oxazolyl)methylene]-(4-nitrobenzene)methanol (3b), 1-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-3,3-dimethyl-1-buten-2-ol (3d) and (E)-1-phenyl-2-((3aR)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-ylidene)ethanone (3f) have been characterised in the solid-state by single crystal X-ray diffraction studies. These data represent the first solid-state structural studies of this class of compounds and details the first synthesis and full characterisation of chiral derivative 3f. All four of these materials are shown to exist in the solid phase in the enamine tautomeric form (e.g., 3a is best described as 2-[4,4-dimethyl-2-oxazolidinylidene]-1-phenylethanone) and it is suggested (NMR, IR) that this isomeric form is likely also retained in solution (e.g., CDCl3) as the more stable isomer. An investigation of the relative gas phase stabilities of the three possible (i.e., the (Z)-enol, keto and enamine) isomers of all five compounds by DFT at the B3LYP/6-311G(d) level of theory confirms the latter as the most stable form. The energy differences between the enamine and keto tautomers have been calculated to be the lowest for derivative 3d. These results are compared and contrasted with the previously reported NMR studies of such compounds which have identified the keto form as being a minor (albeit solution) tautomer. Equilibrium solution tautomer distributions for 3d are found to be solvent dependent. The protonated form of 3a, isolated as the HSO4− salt (i.e.4a), has been further characterised in the solid state by single crystal X-ray diffraction. These data represent the first example of a protonated oxazoline to be structurally elucidated and confirms that upon protonation, the keto (oxazoline) tautomer is the energetically favoured form in the solid-state. This observation is further supported by DFT studies for the gas phase protonated forms of such materials. Further DFT (B3LYP/6-311G(d)) calculations employing the SM8 or SMD solvation models were then applied to address the observed solution isomeric distribution for 3d; these results corroborate the gas phase theoretical treatment and also yield values that predict the higher solution stability of the enamine form as observed, although they fail to account for the existence of the keto form as a minor solution state tautomer. To access the availability of an enol-form, via hypothetical de-protonation to the enolate, compound 3a was treated with hydrated Cu(NO3)2 in EtOH solution. The resulting isolated green-coloured product (5), the first metal derivative of this entire class of ligands, is best described (IR, X-ray diffraction) as a coordinated enolate complex, i.e., Cu(3a-H)2. Complex 5 crystallizes in the P21/c space group with four molecules in the unit cell. The coordination geometry around the formal Cu2+ metal centre is determined to be highly distorted square planar in nature (τ4 = 0.442). TD-DFT is used to give a reasonable explanation for the intensity of the absorbance band observed in the visible region for solutions of 5. These latter experiments strongly suggest that the title class of compounds may have considerable potential as ligands in coordination chemistry and/or metal-mediated catalysis.

 Please wait while we load your content...
Please wait while we load your content...

