
Phosphine, isocyanide, and alkyne reactivity at pentanuclear molybdenum/tungsten–iridium clusters†
Abstract
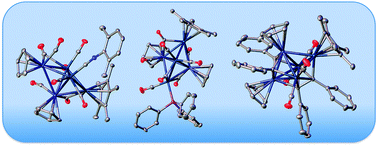
The trigonal bipyramidal clusters M2Ir3(μ-CO)3(CO)6(η5-C5H5)2(η5-C5Me4R) (M = Mo, R = Me 1a, R = H; M = W, R = Me, H) reacted with isocyanides to give ligand substitution products M2Ir3(μ-CO)3(CO)5(CNR′)(η5-C5H5)2(η5-C5Me4R) (M = Mo, R = Me, R′ = C6H3Me2-2,6 3a; M = Mo, R = Me, R′ = tBu 3b), in which core geometry and metal atom locations are maintained, whereas reactions with PPh3 afforded M2Ir3(μ-CO)4(CO)4(PPh3)(η5-C5H5)2(η5-C5Me4R) (M = Mo, R = Me 4a, H 4c; M = W, R = Me 4b, H), with retention of core geometry but with effective site-exchange of the precursors’ apical Mo/W with an equatorial Ir. Similar treatment of trigonal bipyramidal MIr4(μ-CO)3(CO)7(η5-C5H5)(η5-C5Me5) (M = Mo 2a, W 2b) with PPh3 afforded the mono-substitution products MIr4(μ-CO)3(CO)6(PPh3)(η5-C5H5)(η5-C5Me5) (M = Mo 5a; M = W 5b), and further reaction of the molybdenum example 5a with excess PPh3 afforded the bis-substituted cluster MoIr4(μ3-CO)2(μ-CO)2(CO)4(PPh3)2(η5-C5H5)(η5-C5Me5) (6). Reaction of 1a with diphenylacetylene proceeded with alkyne coordination and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C cleavage, affording Mo2Ir3(μ4–η2-PhC2Ph)(μ3-CPh)2(CO)4(η5-C5H5)2(η5-C5Me5) (7a) together with an isomer. Reactions of 2a and 2b with PhCCR afforded MIr4(μ3–η2-PhC2R)(μ3-CO)2(CO)6(η5-C5H5)(η5-C5Me5) (M = Mo, R = Ph 8a; M = W, R = Ph 8b, H; M = W, R = C6H4(C2Ph)-3 9a, C6H4(C2Ph)-4), while addition of 0.5 equivalents of the diynes 1,3-C6H4(C2Ph)2 and 1,4-C6H4(C2Ph)2 to WIr4(μ-CO)3(CO)7(η5-C5H5)(η5-C5Me5) gave the linked clusters [WIr4(CO)8(η5-C5H5)(η5-C5Me5)]2(μ6–η4-PhC2C6H4(C2Ph)-X) (X = 3, 4). The structures of 3a, 4a–4c, 5b, 6, 7a, 8a, 8b and 9a were determined by single-crystal X-ray diffraction studies, establishing the core isomerization of 4, the site selectivity for ligand substitution in 3–6, the alkyne CC dismutation in 7, and the site of alkyne coordination in 7–9. For clusters 3–6, ease of oxidation increases on increasing donor strength of ligand, increasing extent of ligand substitution, replacing Mo by W, and decreasing core Ir content, the Ir-rich clusters 5 and 6 being the most reversible. For clusters 7–9, ease of oxidation diminishes on replacing Mo by W, increasing the Ir content, and proceeding from mono-yne to diyne, although the latter two changes are small. In situ UV-vis-near-IR spectroelectrochemical studies of the (electrochemically reversible) reduction process of 8b were undertaken, the spectra becoming increasingly broad and featureless following reduction. The incorporation of isocyanides, phosphines, or alkyne residues in these pentanuclear clusters all result in an increased ease of oxidation and decreased ease of reduction, and thereby tune the electron richness of the clusters.
C cleavage, affording Mo2Ir3(μ4–η2-PhC2Ph)(μ3-CPh)2(CO)4(η5-C5H5)2(η5-C5Me5) (7a) together with an isomer. Reactions of 2a and 2b with PhCCR afforded MIr4(μ3–η2-PhC2R)(μ3-CO)2(CO)6(η5-C5H5)(η5-C5Me5) (M = Mo, R = Ph 8a; M = W, R = Ph 8b, H; M = W, R = C6H4(C2Ph)-3 9a, C6H4(C2Ph)-4), while addition of 0.5 equivalents of the diynes 1,3-C6H4(C2Ph)2 and 1,4-C6H4(C2Ph)2 to WIr4(μ-CO)3(CO)7(η5-C5H5)(η5-C5Me5) gave the linked clusters [WIr4(CO)8(η5-C5H5)(η5-C5Me5)]2(μ6–η4-PhC2C6H4(C2Ph)-X) (X = 3, 4). The structures of 3a, 4a–4c, 5b, 6, 7a, 8a, 8b and 9a were determined by single-crystal X-ray diffraction studies, establishing the core isomerization of 4, the site selectivity for ligand substitution in 3–6, the alkyne CC dismutation in 7, and the site of alkyne coordination in 7–9. For clusters 3–6, ease of oxidation increases on increasing donor strength of ligand, increasing extent of ligand substitution, replacing Mo by W, and decreasing core Ir content, the Ir-rich clusters 5 and 6 being the most reversible. For clusters 7–9, ease of oxidation diminishes on replacing Mo by W, increasing the Ir content, and proceeding from mono-yne to diyne, although the latter two changes are small. In situ UV-vis-near-IR spectroelectrochemical studies of the (electrochemically reversible) reduction process of 8b were undertaken, the spectra becoming increasingly broad and featureless following reduction. The incorporation of isocyanides, phosphines, or alkyne residues in these pentanuclear clusters all result in an increased ease of oxidation and decreased ease of reduction, and thereby tune the electron richness of the clusters.
 Please wait while we load your content...
Please wait while we load your content...

