
Fast screening of homogeneous catalysis mechanisms using graph-driven searches and approximate quantum chemistry†
 a
and
Scott
Habershon
*a
a
and
Scott
Habershon
*a
Abstract
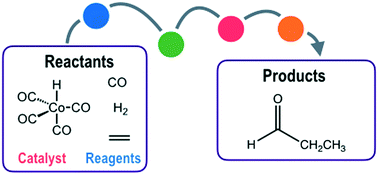
Computational methods for predicting multi-step reaction mechanisms, such as those found in homogeneous catalysis by organometallic complexes, are rapidly emerging as powerful tools to support experimental mechanistic insight. We have recently shown how a graph-driven sampling scheme can be successfully used to propose a series of candidate reaction mechanisms for nanoparticle catalysis; however, identifying the most-likely reaction mechanism amongst this candidate set in an efficient scheme remains a challenge. Here, we show how simple descriptors for each reaction path, calculated using quick semi-empirical quantum chemistry, enable identification of the mechanism, but only if one considers both thermodynamic and kinetic parameters of proposed reaction mechanisms. Successful application to cobalt-catalysed alkene hydroformylation is used to benchmark this strategy, and provides insight into remaining algorithmic challenges.
- This article is part of the themed collection: Celebrating our 2024 Prizewinners
 Please wait while we load your content...
Please wait while we load your content...

