
Origin of distinct structural symmetry of the neopentane cation in the ground electronic state compared to the methane cation
Abstract
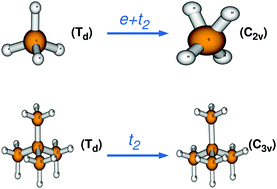
An ab initio quantum dynamics study has been performed to explore the distinct structural symmetry of C(CH3)4+ in the ground electronic state compared to CH4+. Additionally, the underlying details of the highly diffuse and complex vibronic structure of the first photoelectron band of C(CH3)4 have been investigated. Associated potential energy surfaces over the two-dimensional space of nuclear coordinates, subject to the T2 ⊗ (e + t2) Jahn–Teller effect, are established from extensive electronic structure calculations and (then) the nuclear dynamics calculations are done on them via wave packet propagation including the nonadiabatic coupling of the three electronic sheets. The theoretical results are in good agreement with experimental observations. The JT stabilization energies due to T2 ⊗ e, T2 ⊗ t2 and T2 ⊗ (e + t2) distortions in the ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 2T2 electronic manifold of C(CH3)4+ illustrate that the highest stabilization occurs through the T2 ⊗ t2-JT distortion (in the ground state of C(CH3)4+). However, CH4+ gains such maximum stabilization due to T2 ⊗ (e + t2)-JT distortion. From this novel result and applying the epikernel principle, we propose that the structural evolution of C(CH3)4+ from Td to C3v minimum energy configuration occurs via JT active vibrations of t2 symmetry, whereas CH4+ rearranges to the C2v structure through a combination of JT active e and t2 bending vibrations.
2T2 electronic manifold of C(CH3)4+ illustrate that the highest stabilization occurs through the T2 ⊗ t2-JT distortion (in the ground state of C(CH3)4+). However, CH4+ gains such maximum stabilization due to T2 ⊗ (e + t2)-JT distortion. From this novel result and applying the epikernel principle, we propose that the structural evolution of C(CH3)4+ from Td to C3v minimum energy configuration occurs via JT active vibrations of t2 symmetry, whereas CH4+ rearranges to the C2v structure through a combination of JT active e and t2 bending vibrations.
 Please wait while we load your content...
Please wait while we load your content...

