
Theoretical description of efficiency enhancement in DSSCs sensitized by newly synthesized heteroleptic Ru complexes
Abstract
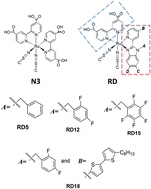
Recently, some new series of heteroleptic ruthenium-based dyes, the so-called RD dyes, were designed and synthesized showing better performances compared to the well-known homoleptic N719. In this work, using the density-functional theory and its time-dependent extension, we have investigated the electronic structure and absorption spectra of these newly synthesized dyes, and compared the results to those of N3 dye to describe the variations of the properties due to the molecular engineering of the ancillary ligand. We have shown that the calculation results of the absorption spectra for these dyes using the PBE0 for the exchange–correlation functional are in better agreement with the experiment than using B3LYP or range-separated CAM-B3LYP. We have also derived a formula based on the DFT and used it to visually describe the level shifts in a solvent. The higher Jsc observed in these new dyes is explained by the fact that here, in contrast to N3, the excitation charge was effectively transferred to the anchoring ligand. Furthermore, we have shown that the difference dipole moment vectors of the ground and excited states can be used to determine the charge-transfer direction in an excitation process. Finally, different electron lifetimes observed in these dyes are explained by investigating the adsorption geometries and the relative orientations of iodine molecules in different “dye⋯I2” complexes.
 Please wait while we load your content...
Please wait while we load your content...

