
Density functional calculations of extended, periodic systems using Coulomb corrected molecular fractionation with conjugated caps method (CC-MFCC)
Abstract
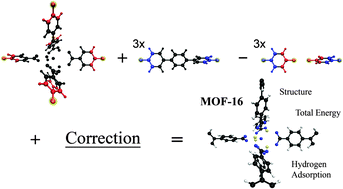
A fragmentation scheme based upon the molecular fractionation with conjugated caps (MFCC) method and derived previously [J. Chem. Phys., 2009, 130, 144104] within the remit of density functional theory (DFT) based on local and semi-local functionals, enables one to perform order-N high-quality DFT calculations on extended systems (e.g. collections of organic molecules) via considering its smaller fragments. Here we discuss in detail a considerably improved method which broadens its applicability to a wider class of extended systems: (i) when each individual fragment is considered, the surrounding part of the entire system is not ignored anymore; instead, it is represented by point charges; (ii) the method is generalised to a system of any complexity enabling studying periodic and porous systems in real space; (iii) an appropriate Coulomb correction term is derived where clear distinction is made between charge densities of the same cap regions appearing in different fragments. Consequently, our correction term turns out to differ substantially from that derived e.g. by Li et al. [J. Chem. Phys. A, 2007, 111(11), 2193]. We also discuss a possibility for the point charges surrounding each fragment to update self-consistently following the calculations of every individual fragment. We examine here a new implementation of our method and its application to a metal–organic framework system. Specifically, we consider the structure of MOF-16 and adsorption of Hydrogen molecules in its pores. Possible ways of improving precision and to further widen up applicability of the method are also discussed.
 Please wait while we load your content...
Please wait while we load your content...

