
Structure prediction of porous organic crystals

Abstract
In this work, we explore the possibility of applying automated crystal structure prediction to reproduce the experimentally identified metastable porous polymorphs. Using our recently developed High-Throughput Organic Crystal Structure Prediction (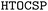 ) framework, we conducted a systematic study on five representative organic crystalline systems including hydrogen-bonded frameworks (HOFs), featured by the presence of significant porosity, in conjunction with different choices of energy models from classical, machine learning force fields, tight binding to density functional theory. Our results suggest that the current structure generation framework, with careful selection of symmetry conditions, is likely to generate rather complex and abundant metastable crystal candidates for porous crystals. In conjunction with the recent advance in universal machine learning force fields, it becomes possible to identify experimental structures as the energetically favorable candidates from a simple energy versus density analysis, thus paving the way for computational design of complex porous materials with the target systems prior to the experimental synthesis and characterization.
) framework, we conducted a systematic study on five representative organic crystalline systems including hydrogen-bonded frameworks (HOFs), featured by the presence of significant porosity, in conjunction with different choices of energy models from classical, machine learning force fields, tight binding to density functional theory. Our results suggest that the current structure generation framework, with careful selection of symmetry conditions, is likely to generate rather complex and abundant metastable crystal candidates for porous crystals. In conjunction with the recent advance in universal machine learning force fields, it becomes possible to identify experimental structures as the energetically favorable candidates from a simple energy versus density analysis, thus paving the way for computational design of complex porous materials with the target systems prior to the experimental synthesis and characterization.

 Please wait while we load your content...
Please wait while we load your content...

