
Near-infrared emitting copper(i) complexes with a pyrazolylpyrimidine ligand: exploring relaxation pathways†

Abstract
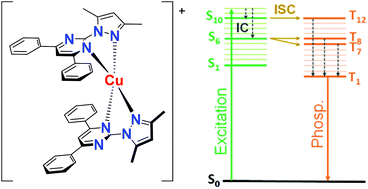
Mononuclear copper(I) complexes [CuL2]I (1), [CuL2]2[Cu2I4]·2MeCN (2) and [CuL2]PF6 (3) with a new chelating pyrazolylpyrimidine ligand, 2-(3,5-dimethyl-1H-pyrazol-1-yl)-4,6-diphenylpyrimidine (L), were synthesized. In the structures of complex cations [CuL2]+, Cu+ ions coordinate two L molecules (N,N-chelating coordination). Extended π-systems of the L molecules in [CuL2]+ favor the formation of paired π–π stacking intramolecular interactions between the pyrimidine and phenyl rings leading to significant distortions of tetrahedral coordination cores, CuN4. The free ligand L demonstrates dual excitation wavelength dependent luminescence in the UV and violet regions, which is attributed to S1 → S0 fluorescence and T1 → S0 phosphorescence with intraligand charge transfer character. The complexes 1–3 demonstrate T1 → S0 phosphorescence in the near-infrared region. Theoretical investigations point to its ligand-to-metal charge transfer (LMCT) origin. Large Stokes shifts of emission (ca. 200 nm) are the result of notable planarizations of CuN4 cores in the T1 state as compared to the S0 state. Spin–orbit coupling computations revealed that the most effective intersystem crossing channels for [CuL2]+ appear in high-lying excited states, while the S1 → T1 transition is unfavourable according to El-Sayed's rule and the energy gap law. Electron-vibration coupling calculations showed that the C–C and C–N stretching vibrations of the pyrimidine and phenyl moieties, the asymmetric Cu–N stretching vibrations and the wagging motions of phenyl rings contribute the most to the non-radiative deactivation of L and [CuL2]+.
 Please wait while we load your content...
Please wait while we load your content...

