
Enantioselective total synthesis of parnafungin A1 and 10a-epi-hirtusneanine†
 *ab
*ab
Abstract
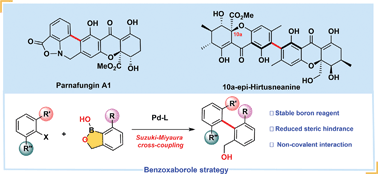
The first and enantioselective total synthesis of the heterodimeric biaryl antifungal natural product parnafungin A1 as well as complex biaryl tetrahydroxanthone 10a-epi-hirtusneanine is accomplished, by employing cross-coupling through the benzoxaborole strategy to construct their sterically hindered biaryl cores. Besides the powerful Suzuki–Miyaura cross-coupling, the synthesis of parnafungin A1 also features a highly diastereoselective oxa-Michael addition to construct a tetrahydroxanthone skeleton, and an effective Zn-mediated reductive cyclization-Mitsunobu sequence to furnish the isoxazolidinone structure. Key innovations in total synthesis of 10a-epi-hirtusneanine include the employment of DTBS protection for functional group manipulation on the tetrahydroxanthone skeleton, stereoselective methylations, and complete reversal of the stereochemistry of the C5-hydroxy group using oxidation/Evans–Saksena reduction, as well as the strategy of preparing both complex tetrahydroxanthone monomers from the same chiral intermediate 25.
 Please wait while we load your content...
Please wait while we load your content...

