
First-principles insights into ammonia decomposition on the MoN(0001) surface
 *a
*a
Abstract
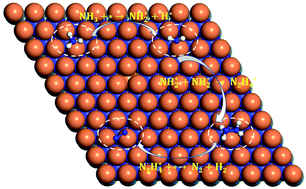
Density functional theory (DFT) and a periodic slab model were used to study the geometric structure, electronic structure and dehydrogenation mechanism of ammonia adsorption on the MoN(0001) surface. The surface energy calculation results indicated that MoN(0001) terminated by N was the most stable surface. The NH3 molecule was preferably adsorbed at the top position through its lone pair electrons, while the most stable adsorption sites of NH2 and H were triple hcp sites, and NH and N were adsorbed at triple fcc sites. The results of transition state calculations showed that the decomposition reaction of ammonia mainly takes place through a step-by-step dehydrogenation mechanism of N2H4, and the compound desorption reaction of nitrogen was the rate limiting step of the whole reaction. The results of electronic structure calculations showed that the N2H4 molecule was adsorbed on the surface through the mixture of the 2Pz orbital of N and the 4dz2 orbital of Mo. With the progress of dehydrogenation, the charge transfer phenomenon became more and more obvious. It could be predicted that the charge transfer between the adsorbate and substrate played an important role in the catalytic process of accelerating the dehydrogenation of NH3.
 Please wait while we load your content...
Please wait while we load your content...

