
Potential energy surface for high-energy N + N2 collisions†
 a
and
Donald G.
Truhlar
*a
a
and
Donald G.
Truhlar
*a
Abstract
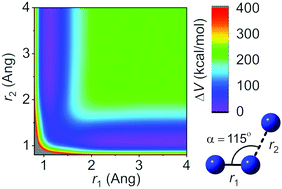
Potential energy surface calculations yield physical insight into the structure of intermediates and the dynamics of molecular collisions, and they are the first step toward molecular simulations that provide physical insight into energy transfer, reaction, and dissociation probabilities. The potential energy surface for high-energy collisions of N2 with N can be used for modeling chemical dynamics and energy transfer in atmospheric shock waves. Here we present an analytic ground-state. (4A′′) potential energy surface for N3 that governs electronically adiabatic collisions of N2(1Σ+g) with N(4S). The fitted surface consists of a pairwise potential based on an accurate diatomic potential energy curve plus a connected permutationally invariant polynomial (PIP) in mixed-exponential-Gaussian bond order variables (MEGs) for the three-body part. The three-body fit is based on multireference complete active space second order perturbation theory (CASPT2) calculations. The quality of the quartet N3 fit is comparable to that for a previous fit of the NO2 potential. We characterize two local minima of N3, two tight transition structures, two van der Waals geometries, and the noncollinear reaction path for the symmetric exchange reaction. The nonreactive approach of an N atom to N2 along the perpendicular bisector is more repulsive than the collinear reproach, but plots of the force on the bond versus the potential energy at the distance of closest approach allow us to infer that vibrational energy transfer should occur much more readily in high-energy collinear collisions than in high-energy perpendicular-bisector collisions.
 Please wait while we load your content...
Please wait while we load your content...

