
Combining artificial intelligence and physics-based modeling to directly assess atomic site stabilities: from sub-nanometer clusters to extended surfaces†

Abstract
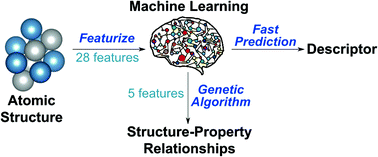
The performance of functional materials is dictated by chemical and structural properties of individual atomic sites. In catalysts, for example, the thermodynamic stability of constituting atomic sites is a key descriptor from which more complex properties, such as molecular adsorption energies and reaction rates, can be derived. In this study, we present a widely applicable machine learning (ML) approach to instantaneously compute the stability of individual atomic sites in structurally and electronically complex nano-materials. Conventionally, we determine such site stabilities using computationally intensive first-principles calculations. With our approach, we predict the stability of atomic sites in sub-nanometer metal clusters of 3–55 atoms with mean absolute errors in the range of 0.11–0.14 eV. To extract physical insights from the ML model, we introduce a genetic algorithm (GA) for feature selection. This algorithm distills the key structural and chemical properties governing the stability of atomic sites in size-selected nanoparticles, allowing for physical interpretability of the models and revealing structure–property relationships. The results of the GA are generally model and materials specific. In the limit of large nanoparticles, the GA identifies features consistent with physics-based models for metal–metal interactions. By combining the ML model with the physics-based model, we predict atomic site stabilities in real time for structures ranging from sub-nanometer metal clusters (3–55 atom) to larger nanoparticles (147 to 309 atoms) to extended surfaces using a physically interpretable framework. Finally, we present a proof of principle showcasing how our approach can determine stable and active nanocatalysts across a generic materials space of structure and composition.
 Please wait while we load your content...
Please wait while we load your content...

