
Theoretical study on the molecular stacking interactions and charge transport properties of triazasumanene crystals – from explanation to prediction†

Abstract
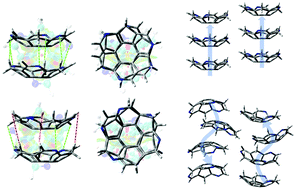
Computational analyses of the solid-state properties of triazasumanene (TAS), a C3-symmetric nitrogen-doped sumanene derivative, were carried out in this work. The present studies are mainly divided into two parts. In the first part, we demonstrated the differences in the interactions of the crystal packing between the racemic and the homochiral structures: the former having perpendicular columnar packing and the latter forming slipped helical packing. Two geometries of the TAS monomer, a theoretically optimized structure under vacuum and an X-ray crystal structure in experiment, were compared. It can be found that it is not the total interaction energy, but the local interactions (mainly the electrostatic interactions) of the molecular dimer that dominate the columnar stacking conformation. The second part involves the investigation of the potential charge transport properties of the crystals according to the semiclassical Marcus theory with the hopping mechanism using the simple dimer model. The charge transfer integrals of the two sets of dimers, racemic and homochiral dimer models, were compared as well. The calculation results show that the TAS racemic crystal was predicted to have an advantage of hole transport properties. The perpendicular columnar stacking of the homochiral conformation should essentially have better charge transport properties than the racemic conformation. It is reasonable to employ the simple dimer model built using optimized monomers under vacuum for the purpose of the prediction of the molecular packing conformation by IES calculation and the charge transport properties of the perpendicular columnar-stacking crystal. Our work provides a simple approach to the deep understanding of the structure–property relationship of bowl-shaped molecular systems in theory. It can help to facilitate the design and preparation of heteroatom-doped sumanene derivatives with perpendicular columnar stacking crystals as novel organic semiconductor materials.
 Please wait while we load your content...
Please wait while we load your content...

