
Searching for double σ- and π-aromaticity in borazine derivatives†

Abstract
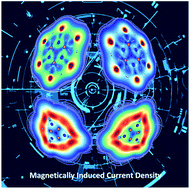
Inspired by the double-aromatic (σ and π) C6H3+, C6I62+, and C6(SePh)62+ ring-shaped compounds, herein we theoretically study their borazine derivative analogues. The systems studied are the cation and dications with formulas B3N3H3+, B3N3Br62+, B3N3I62+, B3N3(SeH)62+, and B3N3(TeH)62+. Our DFT calculations indicate that the ring-shaped planar structures of B3N3H3+, B3N3I62+, and B3N3(TeH)62+ are more stable in the singlet state, while those of B3N3Br62+ and B3N3(SeH)62+ prefer the triplet state. Besides, exploration of the potential energy surface shows that the ring-shaped structure is the putative global minimum only for B3N3I62+. According to chemical bonding analysis, B3N3H3+, B3N3I62+, and B3N3(TeH)62+ have σ and π delocalized bonds. The number of delocalized σ/π electrons is 2/6 for the first, and 10/6 for the second and third, similar to what their carbon analogs exhibit. Finally, the analysis of the magnetically induced current density allows B3N3H3+, B3N3I62+, and B3N3(TeH)62+ to be classified as strongly σ aromatic, and poorly π aromatic compounds.
 Please wait while we load your content...
Please wait while we load your content...

