
An investigation of the metabolic activity, isozyme contribution, species differences and potential drug–drug interactions of PI-103, and the identification of efflux transporters for PI-103-O-glucuronide in HeLa1A9 cells†
 *ab
*ab
Abstract
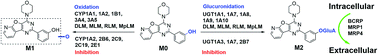
PI-103 is a phosphatidylinositol 3-kinase inhibitor that includes multiple receptor affinity modifications, and it is also a therapeutic drug candidate primarily for human malignant tumors. However, its metabolic fate and potential drug–drug interactions involving human cytochrome P450 (CYP) and UDP-glucuronosyltransferases (UGT) enzymes remain unknown. In this study, our results demonstrated that the intrinsic clearance (CLint) values of oxidated metabolite (M1) in human liver microsomes (HLM) and human intestine microsomes (HIM) were 3.10 and 0.08 μL min−1 mg−1, respectively, while PI-103 underwent efficient glucuronidation with CLint values of 15.59 and 211.04 μL min−1 mg−1 for mono-glucuronide (M2) by HLM and HIM, respectively. Additionally, reaction phenotyping results indicated that CYP1A1 (51.50 μL min−1 mg−1), 1A2 (46.96 μL min−1 mg−1), and UGT1A1 (18.80 μL min−1 mg−1), 1A7 (8.52 μL min−1 mg−1), 1A8 (8.38 μL min−1 mg−1), 1A9 (34.62 μL min−1 mg−1), 1A10 (107.01 μL min−1 mg−1) were the most important contributors for the oxidation and glucuronidation of PI-103. Chemical inhibition assays also suggest that CYP1A2 and UGT1A1, 1A9 play a predominant role in the metabolism of PI-103 in HLM. Significant activity correlations were detected between phenacetin-N-deacetylation and M1 (r = 0.760, p = 0.004) as well as β-estradiol-3-O-glucuronide and M2 (r = 0.589, p = 0.044), and propofol-O-glucuronidation and M2 (r = 0.717, p = 0.009). Furthermore, the metabolism of PI-103 revealed marked species differences, and dogs, rats, mice and mini-pigs were not the appropriate animal models. Gene silencing of breast cancer resistance protein (BCRP) or multidrug resistance-associated protein (MRPs) transporter results indicated that M2 was mainly excreted by BCRP, MRP1 and MRP4 transporters. Moreover, PI-103 displayed broad-spectrum inhibition towards human CYPs and UGTs isozymes with IC50 values ranging from 0.33 to 6.89 μM. Among them, PI-103 showed potent non-competitive inhibitory effects against CYP1A2, 2C19, 2E1 with IC50 and Ki values of less than 1 μM. In addition, PI-103 exhibited moderate non-competitive inhibition against UGT1A7, 2B7, and moderate mixed-type inhibition towards CYP2B6, 2C9 and UGT1A3. Their IC50 and Ki values were 1.16–6.89 and 0.56–5.64 μM, respectively. In contrast, PI-103 could activate the activity of UGT1A4 in a mechanistic two-site model with a Ki value of 13.76 μM. Taken together, PI-103 was subjected to significant hepatic and intestinal metabolism. CYP1A1, 1A2 and UGT1A1, 1A7, 1A8, 1A9, 1A10 were the main contributing isozymes, whereas BCRP, MRP1 and MRP4 contributed most to the efflux excretion of M2. Meanwhile, PI-103 had a potent and broad-spectrum inhibitory effect against human CYPs and UGTs isozymes. These findings could improve understanding of the metabolic fates and efflux transport of PI-103. The inhibited human CYP and UGT activities could trigger harmful DDIs when PI-103 is co-administered with clinical drugs primarily cleared by these CYPs or UGTs isoforms. Additional in vivo studies are required to evaluate the clinical significance of the data presented herein.
 Please wait while we load your content...
Please wait while we load your content...

