
Combined DFT and MD simulation studies of protein stability on imidazolium–water (ImH+Wn) clusters with aromatic amino acids†
 *a
and
*a
and
Abstract
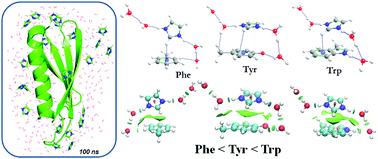
Imidazole (Im) and imidazolium (ImH+) cations play a vital role in the stability of biomolecules in an aqueous environment. The importance of water molecules in the interaction between ImH+ and aromatic amino acids (AAs) is unprecedented. Thus, we have used both quantum mechanical (QM) and molecular dynamics (MD) simulation approaches to understand the nature of interactions between AAs and ImH+ with water molecules. The microscopic studies of AA models and ImH+–water (ImH+Wn, where n = 0–4) clusters were calculated using the M05-2X/6-31+G** method. From DFT calculations, it is observed that the absence of water molecules in ImH+ favors the N–H⋯π interactions (i.e., T-shape) with AAs, whereas the microhydrated clusters of ImH+ prefer to stabilize the stacking conformation. The presence of water-mediated hydrogen bonding (H-bonding) interactions is highly favorable for the stacked confirmation of AAs–ImH+ complexes. This also directly influences the energetics of the cluster. The magnitude of the interaction strength between ImH+Wn and the selected AAs is in the order of Phe < Tyr ∼ Trp. The water-mediated H-bonding interactions act as the main driving force for the stability of π-stacking complexes. Both DFT and MD studies reveal that the ImH+ cation and water molecules play a vital role in the protein stability and also this can intuitively initiate the denaturation process.
 Please wait while we load your content...
Please wait while we load your content...

