
Insight into the active site and reaction mechanism for selective oxidation of methane to methanol using H2O2 on a Rh1/ZrO2 catalyst

Abstract
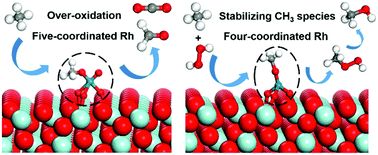
Direct methane conversion into value-added products has become increasingly important. However, it remains a great challenge to effectively activate methane and simultaneously suppress its over-oxidation. In this study, we performed a combined ab initio thermodynamics and DFT+U study to investigate the selective oxidation of methane to methanol on a ZrO2-supported Rh single-atom catalyst. The most preferred local environment of a Rh single atom was proposed according to the ab initio thermodynamics results. The DFT calculation results show that the five-coordinated Rh structure leads to the over-oxidation of CH3 species and thus prevents the formation of methanol. In contrast, the four-coordinated Rh can effectively stabilize the CH3 species by suppressing its further dehydrogenation. This is attributed to the fact that the geometric configuration of CH3 species at the four-coordinated Rh hinders the interaction between H in CH3 species and neighboring O. Two different methanol formation mechanisms at the four-coordinated Rh, namely the direct pathway and the CH3OOH intermediate pathway, were studied. It was found that the four-coordinated Rh facilitates the activation of H2O2 and the formation of CH3OOH, and thus the CH3OOH intermediate pathway plays a dominant role in methanol formation, in which CH3O species reacts with the OH group in H2O2 to form the CH3OOH intermediate and subsequently the deoxygenation of CH3OOH leads to the formation of methanol. This study provides atomic-scale insights into the active site and reaction mechanism for selective oxidation of methane to methanol on Rh1/ZrO2 catalysts.
 Please wait while we load your content...
Please wait while we load your content...

