
Investigating structure-charge transport relationships in thiophene substituted naphthyridine crystalline materials by computational model systems†

Abstract
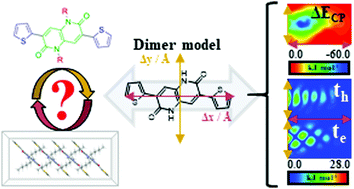
The development of novel π-conjugated charge transfer mediators is at the forefront of current research efforts and interests. Among the plethora of building blocks, diketopyrrolopyrroles have been widely employed, associated to the ease of tailoring their optoelectronic properties by systematic peripheral substitutions. It is somehow of surprise to us that their six-member ring bis-lactam analogues, naphthyridines have been overlooked and reports are scarce and almost solely limited to their use in polymeric materials. Herein we report a comprehensive theoretical analysis of the charge transfer properties of 1,5-naphthyridine-based materials by means of a number of bespoke model systems, further able to quantitatively predict experimental mobility observations. Our results imply that thiophene substituted naphthyridine crystalline materials represent a promising class of organic π-conjugated systems with an experimentally observed ability to self-assemble in the solid state conforming to one dimensional stacking motifs. These highly sought-after charge propagation channels are characterised by large wavefunction overlap and thermal integrity and have as a result the potential to outperform currently exploited alternatives. We anticipate this work to be of interest to materials scientists and hope it will pave the way towards the realisation of novel charge transfer mediators exploiting naphthyridine chemistries.
 Please wait while we load your content...
Please wait while we load your content...

