
Quantum simulation of electronic structure with a transcorrelated Hamiltonian: improved accuracy with a smaller footprint on the quantum computer
 †*a
†*a
Abstract
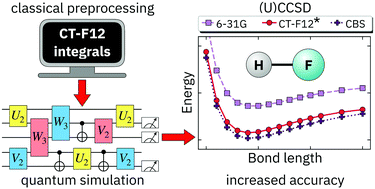
Quantum simulations of electronic structure with a transformed Hamiltonian that includes some electron correlation effects are demonstrated. The transcorrelated Hamiltonian used in this work is efficiently constructed classically, at polynomial cost, by an approximate similarity transformation with an explicitly correlated two-body unitary operator. This Hamiltonian is Hermitian, includes no more than two-particle interactions, and is free of electron–electron singularities. We investigate the effect of such a transformed Hamiltonian on the accuracy and computational cost of quantum simulations by focusing on a widely used solver for the Schrödinger equation, namely the variational quantum eigensolver method, based on the unitary coupled cluster with singles and doubles (q-UCCSD) Ansatz. Nevertheless, the formalism presented here translates straightforwardly to other quantum algorithms for chemistry. Our results demonstrate that a transcorrelated Hamiltonian, paired with extremely compact bases, produces explicitly correlated energies comparable to those from much larger bases. For the chemical species studied here, explicitly correlated energies based on an underlying 6-31G basis had cc-pVTZ quality. The use of the very compact transcorrelated Hamiltonian reduces the number of CNOT gates required to achieve cc-pVTZ quality by up to two orders of magnitude, and the number of qubits by a factor of three.
- This article is part of the themed collections: Quantum computing and quantum information storage: Celebrating the 2022 Nobel Prize in Physics, Quantum Computing and Quantum Information Storage and 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

