
Identifying and explaining vibrational modes of quinacridones via temperature-resolved terahertz spectroscopy: absorption experiments and solid-state density functional theory simulations†

Abstract
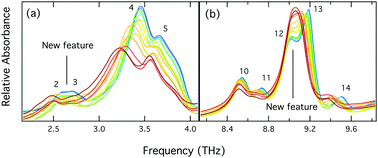
Quinacridone and its substituted analogs are pigments widely used in art and industry. The temperature dependence of the crystal structures of two quinacridone polymorphs (β and γ), along with the common variant 2,9-dimethylquinacridone, were investigated using powder X-ray diffraction and terahertz spectroscopy. These were then compared with solid-state density functional theory simulations of both structures and vibrations. X-ray patterns were collected at eight temperatures in the range 13–298 K and terahertz spectra at fifteen temperatures in the range 20–300 K. Simulations were at absolute zero and at appropriate expansions to model room temperature. It was found that some of the powder X-ray diffraction features in only β-quinacridone (15.7°, 19.7° and 31.2° at 13 K) underwent anomalous shifting with temperature change. We attribute this to the unique coplanar hydrogen bonding pattern of β-quinacridone compared to the other solids, with the unusual diffraction peaks originating from crystallographic planes perpendicular to the a axis intermolecular hydrogen bonds. This observation coincides with a contraction of the a axis with heating and results from its relatively weak N–H⋯O hydrogen bonds and significant C–H⋯H–C repulsions. Associated with this anomalous contraction, for β-quinacridone only spectral peaks are seen to increase in energy with temperature.
 Please wait while we load your content...
Please wait while we load your content...

