
Theoretical characterization of hexagonal 2D Be3N2 monolayers†
 *a
Pablo A.
Denis,
b
*a
Pablo A.
Denis,
b
Abstract
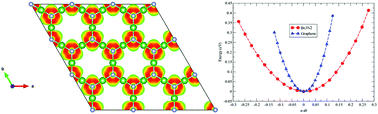
First-principles density functional theory (DFT) calculations are performed to assess the stability, and geometric, mechanical, optical and electronic properties of monolayer graphene-like Be3N2. We find that Be3N2 is a large band gap semiconductor with small electron and hole effective masses, which may promote its use in nanoelectronic devices. Furthermore, its excellent thermal, dynamical, and mechanical stability makes it a material of comparable caliber to that of graphene. In addition, the excellent electrochemical properties of Be3N2 make it a unique material with possible theoretical capacities of 974 mA h g−1 for Li, Na, and K. Moreover, Be3N2 can form bulk graphite-like layered structures with two different configurations, i.e. N2–N1 and N2–Be1. Finally, the derivatives of Be3N2 (Be3N2 nanoribbons) also possess direct band gaps which can be finely tuned to the desired level by geometry and morphology constraints. Based on these fascinating properties, Be3N2 and its derivatives can find a broad range of applications in nanoelectronics and battery technologies.
 Please wait while we load your content...
Please wait while we load your content...

