
 abd
and
A. J.
Logsdail
*a
abd
and
A. J.
Logsdail
*a
Abstract
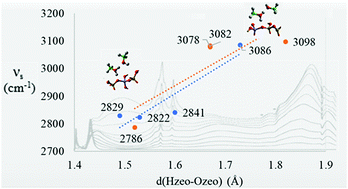
The transformation of methanol-to-hydrocarbons (MTH) has significant potential as a route to synthesise low-cost fuels; however, the initial stages of the zeolite catalysed MTH process are not well understood. Here, we use hybrid quantum- and molecular-mechanical (QM/MM) embedded-cluster simulations to develop our understanding of the interaction between methanol and the zeolite catalysts H-ZSM-5, and for comparison, the larger pore H-Y. Energies and structures, calculated using hybrid-level density functional theory (hybrid-DFT) and higher-level correlated methods, are compared with previous experimental and computational results. We show that hydrogen-bonds between methanol adsorbates, formed through polarizable O–H bonds, substantially influence the adsorption energetics, structural parameters and vibrational frequencies. Our observations are extended by considering polar solvent molecules in the environment, with the presence of both water or methanol around the adsorption site leading to barrier-less transfer of the zeolite proton to an adsorbed methanol, which will significantly influence the reactivity of the adsorbed methanol.
 Please wait while we load your content...
Please wait while we load your content...

