
Ultrafast ring closing of a diarylethene-based photoswitchable nucleoside†

Abstract
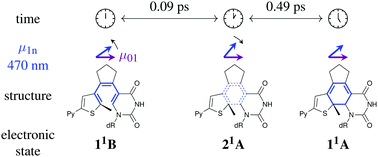
Deoxyuridine nucleosides embodied into diarylethenes form an especial class of photoswitchable compounds that are designed to stack and pair with DNA bases. The molecular geometry can be switched between “open” and “closed” isomers by a pericyclic reaction that affects the stability of the surrounding double helix. This potentially enables light-induced control of DNA hybridization at microscopic resolution. Despite its importance for the optimization of DNA photoswitches, the ultrafast photoisomerization mechanism of these diarylethenes is still not well understood. In this work, femtosecond transient absorption spectroscopy is applied to study the ring closing reaction upon UV excitation with 45 fs pulses. Excited-state absorption decays rapidly and gives rise to the UV-Vis difference spectrum of the “closed” form within ≈15 ps. Time constants of 0.09, 0.49 and 6.6 ps characterize the multimodal dynamics, where a swift recurrence in the signal anisotropy indicates transient population of the intermediate 21A—like state.
 Please wait while we load your content...
Please wait while we load your content...

