
Effects of the c-Si/a-SiO2 interfacial atomic structure on its band alignment: an ab initio study†
 a
a
Abstract
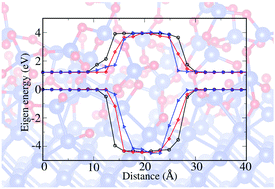
The crystalline-Si/amorphous-SiO2 (c-Si/a-SiO2) interface is an important system used in many applications, ranging from transistors to solar cells. The transition region of the c-Si/a-SiO2 interface plays a critical role in determining the band alignment between the two regions. However, the question of how this interface band offset is affected by the transition region thickness and its local atomic arrangement is yet to be fully investigated. Here, by controlling the parameters of the classical Monte Carlo bond switching algorithm, we have generated the atomic structures of the interfaces with various thicknesses, as well as containing Si at different oxidation states. A hybrid functional method, as shown by our calculations to reproduce the GW and experimental results for bulk Si and SiO2, was used to calculate the electronic structure of the heterojunction. This allowed us to study the correlation between the interface band characterization and its atomic structures. We found that although the systems with different thicknesses showed quite different atomic structures near the transition region, the calculated band offset tended to be the same, unaffected by the details of the interfacial structure. Our band offset calculation agrees well with the experimental measurements. This robustness of the interfacial electronic structure to its interfacial atomic details could be another reason for the success of the c-Si/a-SiO2 interface in Si-based electronic applications. Nevertheless, when a reactive force field is used to generate the a-SiO2 and c-Si/a-SiO2 interfaces, the band offset significantly deviates from the experimental values by about 1 eV.
 Please wait while we load your content...
Please wait while we load your content...

