
Atomic structure of Mg-based metallic glasses from molecular dynamics and neutron diffraction

Abstract
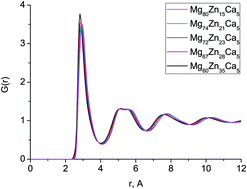
We use a combination of classical molecular dynamics simulation and neutron diffraction to identify the atomic structure of five different Mg–Zn–Ca bulk metallic glasses, covering a range of compositions with substantially different behaviour when implanted in vitro. There is very good agreement between the structures obtained from computer simulation and those found experimentally. Bond lengths and the total correlation function do not change significantly with composition. The zinc and calcium bonding shows differences between composition: the distribution of Zn–Ca bond lengths becomes narrower with increasing Zn content, and the preference for Zn and Ca to avoid bonding to themselves or each other becomes less strong, and, for Zn–Ca, transforms into a positive preference to bond to each other. This transition occurs at about the same Zn content at which the behaviour on implantation changes, hinting at a possible structural connection. A very broad distribution of Voronoi polyhedra are also found, and this distribution broadens with increasing Zn content. The efficient cluster packing model, which is often used to describe the structure of bulk metallic glasses, was found not to describe these systems well.
 Please wait while we load your content...
Please wait while we load your content...

