
Covalent tethering of fragments for covalent probe discovery†
Abstract
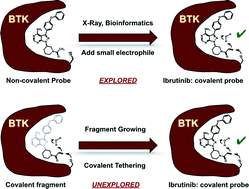
Covalent probes and drugs have found widespread use as research tools and clinical agents. Covalent probes are useful because of their increased intracellular potency and because covalent labeling of cellular proteins can be tracked using click chemistry. Covalent drugs, on the other hand, can overcome drug resistance toward their reversible counterparts. The discovery of covalent probes and drugs usually follows two trajectories: covalent natural products and their analogues are used directly as covalent probes or drugs; or alternatively, a non-covalent probe is equipped with a reactive group and converted into a covalent probe. In both cases, there is a need to either have a natural product or a potent non-covalent scaffold. The alternative approach to discover covalent probes is to start with a drug-like fragment that already has an electrophile, and then grow the fragment into a potent lead compound. In this approach, the electrophilic fragment will react covalently with the target protein, and therefore the initial weak binding of the fragment can be amplified over time and detected using mass spectrometry. With this approach the surface of the protein can be interrogated with a library of covalent fragments to identify covalent drug binding sites. One challenge with this approach is the danger of non-specific covalent labeling of proteins with covalent fragments. The second challenge is the risk of selecting the most reactive fragment rather than the best binder if the covalent fragments are screened in mixtures. This review will highlight how covalent tethering was developed, its current state, and its future.
 Please wait while we load your content...
Please wait while we load your content...