
First-principles study of pressure-induced structural phase transitions in MnF2
 *a
A. H.
Romero,
be
*a
A. H.
Romero,
be
Abstract
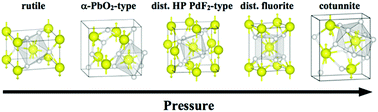
In this work we report a complete structural and magnetic characterization of crystalline MnF2 under pressure obtained using first principle calculations. Density functional theory was used as the theoretical framework, within the generalized gradient approximation plus the Hubbard formalism (GGA+U) necessary to describe the strong correlations present in this material. The vibrational, the magnetic exchange couplings and the structural characterization of MnF2 in the rutile ground state structure and potential high pressure phases are reported. The quasiharmonic approximation has been used to obtain the free energy, which at the same time is used to evaluate the different structural transitions at 300 K. Based on previous theoretical and experimental studies on AF2 compounds, ten different structural candidates were considered for the high pressure regime, which led us to propose a path for the MnF2 structural transitions under pressure. As experimental pressure settings can lead to non-hydrostatic conditions, we consider hydrostatic and non-hydrostatic strains in our calculations. According to our results we found the following sequence for the pressure-driven structural phase transition in MnF2: rutile (P42/mnm) → α-PbO2-type (Pbcn) → dist. HP PdF2-type (Pbca) → dist. fluorite (I4/mmm) → cotunnite (Pnma). This structural path is correlated with other phase transitions reported on other metal rutile fluorides. In particular, we found that our proposed structural phase transition sequence offers an explanation of the different paths observed in the literature by taking into account the role of the hydrostatic conditions. In order to get a deep understanding of the modifications of MnF2 under pressure, we have analyzed the pressure evolution of the structural, vibrational, electronic, and magnetic properties for rutile and for each of the high pressure phases.
 Please wait while we load your content...
Please wait while we load your content...

