
Nuclear size effects in vibrational spectra
Abstract
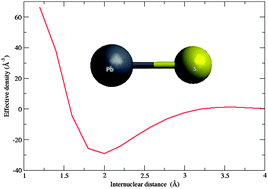
We present a theoretical study of nuclear volume in the rovibrational spectra of diatomic molecules which is an extension of a previous study restricted to rotational spectra [Chem. Phys., 2012, 401, 103]. We provide a new derivation for the electron–nucleus electrostatic interaction energy which is basically independent of the choice of model for the nuclear charge distribution. Starting from this expression we derive expressions for the electronic, rotational and vibrational field shift parameters in terms of effective electron density and its first and second derivatives with respect to internuclear distance. The effective density is often approximated by the contact density, but we demonstrate that this leads to errors on the order of 10% and is furthermore not necessary since the contact and effective densities can be obtained at the same computational cost. We calculate the field shift parameters at the 4-component relativistic coupled-cluster singles-and-doubles level and find that our results confirm the experimental findings of Tiemann and co-workers [Chem. Phys., 1982, 68(21), 1982, Ber. Bunsenges. Phys. Chem., 1982, 86, 821], whereas we find no theoretical justification for a scaling factor introduced in later work [Chem. Phys., 1985, 93, 349]. For lead sulfide we study the effective density as a function of internuclear distance and find a minimum some 0.2 Å inside the equilibrium bond distance. We also discuss Bigeleisen–Goeppert-Mayer theory of isotope fractionation in light of our results.
 Please wait while we load your content...
Please wait while we load your content...

