
Density functional theory study of oxygen reduction reaction on Pt/Pd3Al(111) alloy electrocatalyst
Abstract
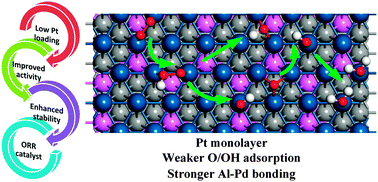
Developing efficient catalysts for the oxygen reduction reaction (ORR) to reduce cathode Pt loading without sacrificing the performance has been under intensive research. Herein, by using density functional theory calculations, the activity and stability of a Pt monolayer supported on Pd3Al(111) as the ORR catalyst have been systematically studied. The simulations demonstrate that due to alloying, the ORR intermediates bind weakly on Pt/Pd3Al(111) with optimal adsorption energy of O and OH. By considering the elemental ORR steps, the ORR mechanism is predicted to be an OOH dissociation mechanism. The rate determining step is OOH dissociation with a reaction barrier of 0.37 eV, lower than the corresponding value on Pt/Pt3Al(111) and Pt(111), indicating the superior activity of Pt/Pd3Al(111). Even considering the unfeasible H adsorption under high potential, the ORR mechanism on Pt/Pd3Al(111) would proceed via O2 hydration, OOH hydration, H2O formation, and H2O desorption, indicating a good ORR electrocatalyst. Furthermore, stability was evaluated by calculating the alloy formation energy and the electrochemical potential shift of surface Pt dissolution. The exceptionally negative alloy formation energy of Pd3Al and the positive dissolution potential shift of the surface Pt atoms show the enhanced durability of Pt/Pd3Al(111). The improved activity, in combination with its enhanced stability, makes the novel ternary alloy electrocatalyst very promising for development of new cathode catalysts for fuel cells.
 Please wait while we load your content...
Please wait while we load your content...

