
Rationally designed donor–acceptor scheme based molecules for applications in opto-electronic devices
Abstract
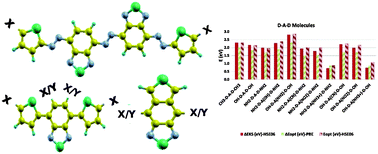
Several donor (D)–acceptor (A) based molecules are rationally designed by adopting three different schemes in which the conjugation length, strength of the donor and acceptor moieties, and planarity of the molecules are varied. These variations are made by introducing a π-conjugated linkage unit, terminating the ends of the moieties by different electron donating and accepting functional groups, and fusing the donor and acceptor moieties, respectively. Our DFT and TDDFT based calculations reveal that using the above-mentioned design schemes, the electronic and optical properties of the D–A based molecules can be largely tuned. While introduction of a linkage and fusing of moieties enhance the π–π interaction, addition of electron donating groups (–CH3, –OH, and –NH2) and electron accepting groups (–CF3, –CN, –NO2, and –NH3+) varies the strength of the donor and acceptor moieties. These factors lead to modulation of the HOMO and LUMO energy levels and facilitate the engineering of the HOMO–LUMO gap and the optical gap over a wide range of ∼0.7–3.7 eV. Moreover, on the basis of calculated ionization potential and reorganization energy, most of the investigated molecules are predicted to be air stable and to exhibit high electron mobility, with the possibility of the presence of ambipolar characteristics in a few of them. The results of our calculations not only demonstrate the examined molecules to be the potential materials for organic opto-electronic devices, but also establish an understanding of the composition–structure–property correlation, which will provide guidelines for designing and synthesizing new materials of choice.
 Please wait while we load your content...
Please wait while we load your content...

