
Modulating intramolecular P⋯N pnictogen interactions†

Abstract
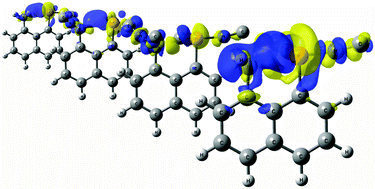
A computational study of the intramolecular pnictogen bond in 8-phosphinonaphthalen-1-amine derivatives (1-NX2, 8-PX2 with X = H, F, Cl, Br, CH3, CN and NC), proton sponge analogues, has been carried out to determine their structural and geometric parameters, interaction energies and electronic properties such as the electron density of the intramolecular interaction. Our results show that substitution of H atoms in the PH2 group by electron withdrawing groups on the Lewis acid moiety strengthens the P⋯N pnictogen bond, evidenced by the increasing electron density values at the bond critical point and by shorter distances. However, substitutions on the Lewis base moiety (NX2) show weaker P⋯N interactions than when the substitution is done on the Lewis acid counterpart (PX2). Nevertheless, in all cases, pnictogen bonds are enhanced upon substitution with respect to the parent 1-NH2, 8-PH2 system. Second-order orbital interaction energies, electron density maps, electron delocalization functions and charge transfer corroborate the evolution of the P⋯N strength upon substitution.
 Please wait while we load your content...
Please wait while we load your content...

