
Quantitative assessment of the carbocation/carbene character of the gold–carbene bond†

Abstract
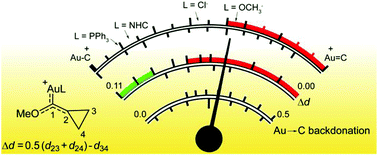
The geometric perturbation of the cyclopropyl ring in [LAu(S)]n+ (S = cyclopropyl(methoxy)carbene) complexes has been recently proposed as an indirect experimental probe of the [LAu]n+ electron-donating power, but experimental data are available only for a phosphine ligand [Brooner et al., Chem. Commun., 2014, 50, 2420, L = P(t-Bu)2(o-biphenyl)]. We broaden the study through DFT geometry optimization of a large number of systems, including anionic, neutral and cationic ligands. We combine these results with the accurate calculation, through charge displacement analysis, of the Dewar–Chatt–Duncanson components of the Au–carbene bond. The results demonstrate a linear correlation between the distortion of the cyclopropyl ring (Δd) and the Au → C π back-donation, which enables us to confidently estimate back-donation from a simple geometry optimization or, when available, from experimental data such as X-ray crystal structures. Consequently, Δd can be reliably used to quantitatively determine the position of each system in the continuum between the carbocationic and carbene extremes and the percentage of back-donation that S is able to accept (Pback). In particular, Pback results to be vanishing with cationic ligands, between 18 and 27% with neutral phosphines and carbenes and around 50% with anionic ligands. Finally, we study the effect of the heteroatom on the substrate, showing that the absolute value of the back-donation is enhanced by around 25% when the methoxy is substituted by a methyl group. Despite this, since the absence of the heteroatom also enhances the maximum capacity of the carbene to accept back-donation, the position of the systems in the continuum moves only slightly toward the carbene end.
 Please wait while we load your content...
Please wait while we load your content...

