
Asymmetric bifurcated halogen bonds†
Abstract
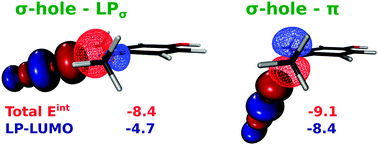
Halogen bonding (XB) is being extensively explored for its potential use in advanced materials and drug design. Despite significant progress in describing this interaction by theoretical and experimental methods, the chemical nature remains somewhat elusive, and it seems to vary with the selected system. In this work we present a detailed DFT analysis of three-center asymmetric halogen bond (XB) formed between dihalogen molecules and variously 4-substituted 1,2-dimethoxybenzene. The energy decomposition, orbital, and electron density analyses suggest that the contribution of electrostatic stabilization is comparable with that of non-electrostatic factors. Both terms increase parallel with increasing negative charge of the electron donor molecule in our model systems. Depending on the orientation of the dihalogen molecules, this bifurcated interaction may be classified as ‘σ-hole – lone pair’ or ‘σ-hole – π’ halogen bonds. Arrangement of the XB investigated here deviates significantly from a recent IUPAC definition of XB and, in analogy to the hydrogen bonding, the term bifurcated halogen bond (BXB) seems to be appropriate for this type of interaction.
 Please wait while we load your content...
Please wait while we load your content...

