
Simple, yet powerful methodologies for conformational sampling of proteins
Abstract
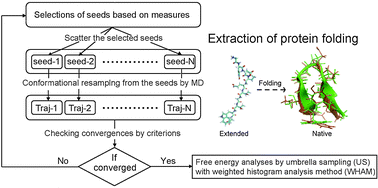
Several biological functions, such as molecular recognition, enzyme catalysis, signal transduction, allosteric regulation, and protein folding, are strongly related to conformational transitions of proteins. These conformational transitions are generally induced as slow dynamics upon collective motions, including biologically relevant large-amplitude fluctuations of proteins. Although molecular dynamics (MD) simulation has become a powerful tool for extracting conformational transitions of proteins, it might still be difficult to reach time scales of the biological functions because the accessible time scales of MD simulations are far from biological time scales, even if straightforward conventional MD (CMD) simulations using massively parallel computers are employed. Thus, it is desirable to develop efficient methods to achieve canonical ensembles with low computational costs. From this perspective, we review several enhanced conformational sampling techniques of biomolecules developed by us. In our methods, multiple independent short-time MD simulations are employed instead of single straightforward long-time CMD simulations. Our basic strategy is as follows: (i) selection of initial seeds (initial structures) for the conformational sampling in restarting MD simulations. Here, the seeds should be selected as candidates with high potential to transit. (ii) Resampling from the selected seeds by initializing velocities in restarting short-time MD simulations. A cycle of these simple protocols might drastically promote the conformational transitions of biomolecules. (iii) Once reactive trajectories extracted from the cycles of short-time MD simulations are obtained, a free energy profile is evaluated by means of umbrella sampling (US) techniques with the weighted histogram analysis method (WHAM) as a post-processing technique. For the selection of the initial seeds, we proposed four different choices: (1) Parallel CaScade molecular dynamics (PaCS-MD), (2) Fluctuation Flooding Method (FFM), (3) Outlier FLOODing (OFLOOD) method, and (4) TaBoo SeArch (TBSA) method. We demonstrate applications of our methods to several biological systems, such as domain motions of proteins with large-amplitude fluctuations, conformational transitions upon ligand binding, and protein folding/refolding to native structures of proteins. Finally, we show the conformational sampling efficiencies of our methods compared with those by CMD simulations and other previously developed enhanced conformational sampling methods.
 Please wait while we load your content...
Please wait while we load your content...

