
Solvation dynamics and energetics of intramolecular hydride transfer reactions in biomass conversion†
Abstract
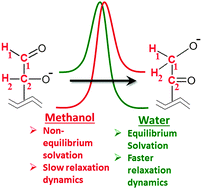
Hydride transfer changes the charge structure of the reactant and thus, may induce reorientation/reorganization of solvent molecules. This solvent reorganization may in turn alter the energetics of the reaction. In the present work, we investigate the intramolecular hydride transfer by taking Lewis acid catalyzed glucose to fructose isomerization as an example. The C2–C1 hydride transfer is the rate limiting step in this reaction. Water and methanol are used as solvents and hydride transfer is simulated in the presence of explicit solvent molecules, treated quantum mechanically and at a finite temperature, using Car–Parrinello molecular dynamics (CPMD) and metadynamics. Activation free energy barrier for hydride transfer in methanol is found to be 50 kJ mol−1 higher than that in water. In contrast, in density functional theory calculations, using an implicit solvent environment, the barriers are almost identical. Analysis of solvent dynamics and electronic polarization along the molecular dynamics trajectory and the results of CPMD-metadynamics simulation of the hydride transfer process in the absence of any solvent suggest that higher barrier in methanol is a result of non-equilibrium solvation. Methanol undergoes electronic polarization during the hydride transfer step. However, its molecular orientational relaxation is a much slower process that takes place after the hydride transfer, over an extended timescale. This results in non-equilibrium solvation. Water, on the other hand, does not undergo significant electronic polarization and thus, has to undergo minimal molecular reorientation to provide near equilibrium solvation to the transition state and an improved equilibrium solvation to the post hydride shift product state. Hence, the hydride transfer step is also observed to be exergonic in water and endergonic in methanol. The aforementioned explanation is juxtaposed to enzyme catalyzed charge transfer reactions, where the enhanced solvation of the transition and product states by enzymes, due to electrostatic interactions, reduces the activation free energy barrier and the free energy change of the reaction. Similarly, we suggest that, in the intramolecular hydride shift, improved solvation of the transition state and of the product state by water is achieved due to minimal polarization and reorientation, and (near) equilibrium solvation.
 Please wait while we load your content...
Please wait while we load your content...

