
Proton transfer in a short hydrogen bond caused by solvation shell fluctuations: an ab initio MD and NMR/UV study of an (OHO)− bonded system
 *ae
*ae
Abstract
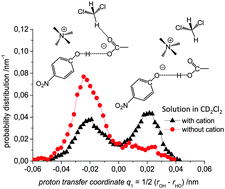
We present a joint experimental and quantum chemical study on the influence of solvent dynamics on the protonation equilibrium in a strongly hydrogen bonded phenol–acetate complex in CD2Cl2. Particular attention is given to the correlation of the proton position distribution with the internal conformation of the complex itself and with fluctuations of the aprotic solvent. Specifically, we have focused on a complex formed by 4-nitrophenol and tetraalkylammonium–acetate in CD2Cl2. Experimentally we have used combined low-temperature 1H and 13C NMR and UV-vis spectroscopy and showed that a very strong OHO hydrogen bond is formed with proton tautomerism (PhOH⋯−OAc and PhO−⋯HOAc forms, both strongly hydrogen bonded). Computationally, we have employed ab initio molecular dynamics (70 and 71 solvent molecules, with and without the presence of a counter-cation, respectively). We demonstrate that the relative motion of the counter-cation and the “free” carbonyl group of the acid plays the major role in the OHO bond geometry and causes proton “jumps”, i.e. interconversion of PhOH⋯−OAc and PhO−⋯HOAc tautomers. Weak H-bonds between CH(CD) groups of the solvent and the oxygen atom of carbonyl stabilize the PhOH⋯−OAc type of structures. Breaking of CH⋯O bonds shifts the equilibrium towards PhO−⋯HOAc form.
 Please wait while we load your content...
Please wait while we load your content...

