
A questionable excited-state double-proton transfer mechanism for 3-hydroxyisoquinoline
Abstract
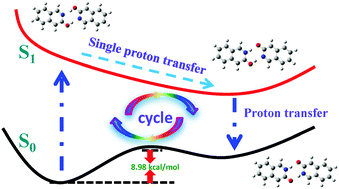
Two excited state proton transfer mechanisms of 3-hydroxyisoquinoline (3HIQ) in cyclohexane and acetic acid (ACID) were investigated based on the time-dependent density functional theory (TDDFT), suggesting a different double-proton transfer mechanism from the one proposed previously (J. Phys. Chem. B, 1998, 102, 1053). Instead of the formation of keto–enol complexes for 3HIQ self-association in cyclohexane, our theoretical results predicted that 3HIQ self-association exists in two forms: the normal form (enol/enol) and the tautomer form (keto/keto) in cyclohexane. A high barrier (37.023 kcal mol−1) between the 3HIQ enol monomer and 3HIQ keto monomer form indicated that the 3HIQ keto monomer in the ground state should not exist. In addition, the constructed potential energy surfaces of the ground state and excited state have been used to explain the proton transfer process. Upon optical excitation, the enol/enol form is excited to the first excited state, then transfers one proton, in turn, transition to the ground state to transfer another proton. A relatively low barrier (8.98 kcal mol−1) demonstrates two stable structures in the ground state. In view of the acetic acid solvent effect, two protons of 3HIQ/ACID transfer along the dihydrogen bonds in the first excited state, which is a different transfer mechanism to 3HIQ self-association. In addition, the proton transfer process provides a possible explanation for the fluorescence quenching observed.
 Please wait while we load your content...
Please wait while we load your content...

