
17O NMR chemical shifts in oxometalates: from the simplest monometallic species to mixed-metal polyoxometalates†
Abstract
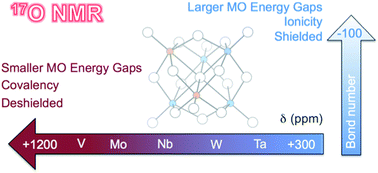
We report a theoretical analysis on 17O NMR chemical shifts for a family of prototypical polyoxometalate anions. The huge diversity of structures and compositions in this family of oxometalates provides a unique resource for evaluating the influence of the metal type and connectivity over the resonance of 17O nuclei. For a set of 75 signals, we show that DFT calculations performed with the GGA-type OPBE functional, including spin–orbit and scaling corrections, provide a mean absolute error <30 ppm, a small value considering that the range of δ(17O) values in these systems is ∼1200 ppm. For terminal M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxygens, the chemical shifts primarily depend on the energy gap between π*M–O and σM–O orbitals. When M is in its highest oxidation state, the energy of π*M–O increases as we replace M going to the left and down in the periodic table. Consequently, we must expect large energy gaps and upfield shifts for O atoms linked to more electropositive ions. Although there is not a direct relationship between δ(17O) and the negative charge of the oxygen, it is not entirely wrong to correlate atomic charge and chemical shift because the ionicity of the M–O bond, the orbital energy gap and the charge density of oxygen are related. The 17O NMR chemical shifts move upfield with an increasing number of bound metal ions because of the larger energy gap in the involved orbitals. Finally, we explored the effect of protonation on δ(17O) in oxometalates and demonstrated that 17O NMR can be a powerful tool to identify the site(s) of protonation at low pH.
O oxygens, the chemical shifts primarily depend on the energy gap between π*M–O and σM–O orbitals. When M is in its highest oxidation state, the energy of π*M–O increases as we replace M going to the left and down in the periodic table. Consequently, we must expect large energy gaps and upfield shifts for O atoms linked to more electropositive ions. Although there is not a direct relationship between δ(17O) and the negative charge of the oxygen, it is not entirely wrong to correlate atomic charge and chemical shift because the ionicity of the M–O bond, the orbital energy gap and the charge density of oxygen are related. The 17O NMR chemical shifts move upfield with an increasing number of bound metal ions because of the larger energy gap in the involved orbitals. Finally, we explored the effect of protonation on δ(17O) in oxometalates and demonstrated that 17O NMR can be a powerful tool to identify the site(s) of protonation at low pH.
 Please wait while we load your content...
Please wait while we load your content...

