
Deciphering global signal features of high-throughput array data from cancers†
Abstract
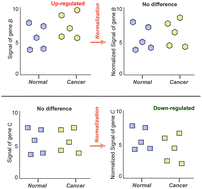
Normalization of array data relies on the assumption that most genes are not altered, which means that the signals for different samples should be scaled to have similar median or average values. However, accumulating evidence suggests that gene expression could be widely up-regulated in cancers. Our previous results and subsequent findings have shown that violation of the assumption led to erroneous interpretation of microarray data. To decipher the global signal features of microarray data from cancer samples, we empirically evaluated a large collection of gene and miRNA expression profiles and copy-number variation arrays. Our results showed that, at the transcriptomic level, genes and miRNAs are widely over-expressed in a large proportion of cancers. In contrast, at the genomic level, global raw signal intensities for methylation and copy number variation show negligible differences between cancer and normal samples. These results force us to re-evaluate the proper use of normalization procedures under different experimental conditions and for different array platforms.
 Please wait while we load your content...
Please wait while we load your content...