
A new exchange–correlation functional free of delocalization and static correlation errors†
Abstract
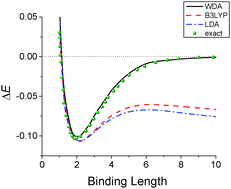
Predicting the correct binding curves of H2+ and H2 systems presents a great challenge in current applications of electronic density functional theory. Here we report a new functional for the exchange–correlation energy based on the weighted density approximation and the classical mapping method. With the exact sum rule for the exchange–correlation hole and accurate correlation functions of uniform electrons as the input, the new functional is free of delocalization and static correlation errors. It yields the exact results for any one-electron systems and the correct asymptotic limit of the binding energy between hydrogen atoms.
 Please wait while we load your content...
Please wait while we load your content...

