
The calculation of 29Si NMR chemical shifts of tetracoordinated silicon compounds in the gas phase and in solution†
Abstract
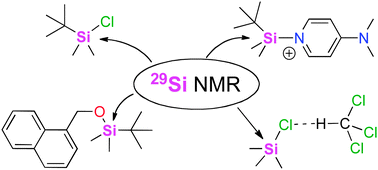
Aiming at the identification of an efficient computational protocol for the accurate NMR assessment of organosilanes in low-polarity organic solvents, 29Si NMR chemical shifts of a selected set of such species relevant in organic synthesis have been calculated relative to tetramethylsilane (TMS, 1) using selected density functional and perturbation theory methods. Satisfactory results are obtained when using triple zeta quality basis sets such as IGLO-III. Solvent effects impact the calculated results through both, changes in substrate geometry as well as changes in the actual shieldings. Spin–orbit (SO) corrections are required for systems carrying more than one chlorine atom directly bonded to silicon. Best overall results are obtained using gas phase geometries optimized at MPW1K/6-31+G(d) level in combination with shielding calculations performed at MPW1K/IGLO-III level in the presence of the PCM continuum solvation model.
 Please wait while we load your content...
Please wait while we load your content...

