
Shock initiated thermal and chemical responses of HMX crystal from ReaxFF molecular dynamics simulation†
Abstract
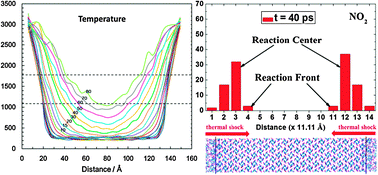
To gain an atomistic-level understanding of the thermal and chemical responses of condensed energetic materials under thermal shock, we developed a thermal shock reactive dynamics (TS-RD) computational protocol using molecular dynamics simulation coupled with ReaxFF force field. β-Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX) was selected as a a target explosive due to its wide usage in the military and industry. The results show that a thermal shock initiated by a large temperature gradient between the “hot” region and the “cold” region results in thermal expansion of the particles and induces a thermal–mechanical wave propagating back and forth in the system with an averaged velocity of 3.32 km s−1. Heat propagating along the direction of thermal shock leads to a temperature increment of the system and thus chemical reaction initiation. Applying a continuum reactive heat conduction model combined with the temperature distribution obtained from the RD simulation, a heat conduction coefficient is derived as 0.80 W m−1 K−1. The chemical reaction mechanisms during thermal shock were analyzed, showing that the reaction is triggered by N–NO2 bond breaking followed by HONO elimination and ring fission. The propagation rates of the reaction front and reaction center are obtained to be 0.069 and 0.038 km s−1, based on the time and spatial distribution of NO2. The pressure effect on the thermal shock was also investigated by employing uniaxial compression before the thermal shock. We find that compression significantly accelerates thermal–mechanical wave propagation and heat conduction, resulting in higher temperature and more excited molecules and thus earlier initiation and faster propagation of chemical reactions.
 Please wait while we load your content...
Please wait while we load your content...

