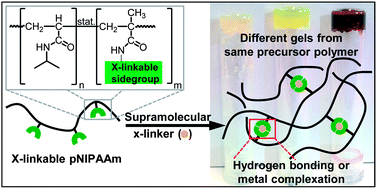
Supramolecular polymer gels are swollen networks of macromolecules interconnected by transient, non-covalent bonds; they form an extraordinarily useful class of soft, stimuli-sensitive materials. To optimize the use of supramolecular polymer gels in applications, their physical and chemical properties must be understood. This understanding is ideally achieved using model systems that allow the type and strength of supramolecular chain crosslinking to be varied to a great extent without concurrent alteration of the properties of the covalent polymer backbones. We introduce a system that provides these requirements. We use linear chains of electrophilic methacryl-succinimidyl (MASI) modified poly(N-isopropylacrylamide) (pNIPAAm). These polymers can be modified in a modular fashion by replacing their electrophilic MASI units by nucleophilic amine-functionalized derivatives of custom, supramolecular crosslinkable functionalities. We follow this approach and prepare a set of pNIPAAm polymers that consist of exactly the same polymer backbone functionalized with different types of crosslinkable sidegroups. These polymers are then crosslinked by addition of low molecular weight linkers that are complementary to the motifs on the polymer. We use multiple hydrogen bonding based on diaminotriazine and maleimide, cyanuric acid and Hamilton wedges, and diaminotriazine and cyanuric acid; we also use metal complexation based on terpyridine and different metal salts. This approach creates supramolecular networks of greatly varying rheological properties, from low viscous liquids to elastic gels, each showing consistent and quantitative correlation between the gel mechanical properties and the binding strength of the respective constituent supramolecular crosslinking motifs. Exploiting the good solubility of the common pNIPAAm backbone polymer in a variety of solvents allows these networks to be prepared and studied in different media with unprecedented consistency and flexibility.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


