
Integrated X-ray photoelectron spectroscopy and DFT characterization of benzene adsorption on Pt(111), Pt(355) and Pt(322) surfaces†
Abstract
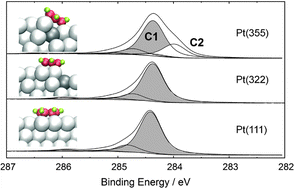
We systematically investigate the adsorption of benzene on Pt(111), Pt(355) and Pt(322) surfaces by high-resolution X-ray photoelectron spectroscopy (XPS) and first-principle calculations based on density functional theory (DFT), including van der Waals corrections. By comparing the adsorption energies at 1/9, 1/16 and 1/25 ML on Pt(111), we find significant lateral interactions exist between the benzene molecules at 1/9 ML. The adsorption behavior on Pt(355) and Pt(322) is very different. While on Pt(355) a step species is clearly identified in the C 1s spectra at low coverages followed by occupation of a terrace species at high coverages, no evidence for a step species is found on Pt(322). These different adsorption sites are confirmed by extensive DFT calculations, where the most favorable adsorption configurations on Pt(355) and Pt(322) are also found to vary: a highly distorted across the step molecule is found on Pt(355) while a less distorted configuration adjacent to the step molecule is deduced for Pt(322). The theoretically proposed C 1s core level binding energy shifts between these most favorable configurations and the terrace species are found to correlate well with experiment: for Pt(355), two adsorbate states are found, separated by ∼0.4 eV in XPS and 0.3 eV in the calculations, in contrast to only one state on Pt(322).
 Please wait while we load your content...
Please wait while we load your content...

